
Sickle Cell Disease (SCD) is a group of disorders that adversely affect the body’s production of hemoglobin, the component in red blood cells that delivers oxygen throughout the body.1 Individuals with these disorders possess atypical hemoglobin molecules, called hemoglobin “S,” which can distort the red blood cell morphology into a sickle, or crescent, shape. Multiple variants of the hemoglobin S molecule exist, and each genetic sub-type is characterized by different prognostic indications.
Graphic 1. Depiction of a Sickle Cell
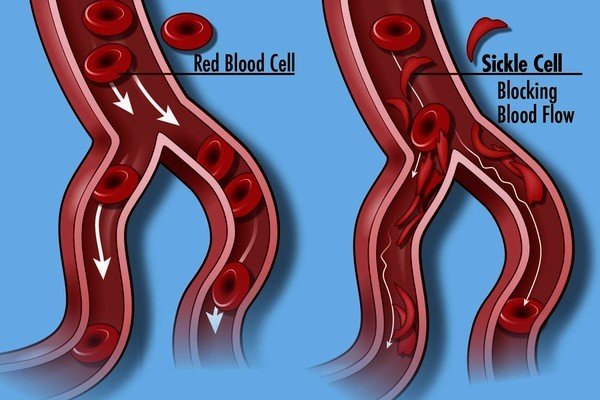
Sickle cell disease (SCD) is often diagnosed at birth following newborn screening or following severe and / or persistent infections. Symptoms of sickle cell disorders include anemia (low red blood cell counts) as a result of the sickle cells dying prematurely, repeat infections, and periodic episodes of pain (i.e., pain crisis). The severity of symptoms varies from person to person ranging from mild to severe. Refer to Table 1. Sickle Cell Disease Symptoms for additional symptoms associated with SCD. Hematopoietic cell transplant (HCT) is currently the major curative treatment for Sickle Cell Disease (SCD).
Table 1. Sickle Cell Disease Symptoms2
| Common SCD Symptoms |
|---|
| 100% of people have these symptoms: |
| Chronic hemolytic anemia |
| 80 – 99% of people have these symptoms: |
| Recurrent infections |
| 30 – 79% of people have these symptoms: |
| Abnormality of the spleen |
| Avascular necrosis |
| Chest pain |
| Iron deficiency |
| Leukocytosis |
| Osteomyelitis |
| Osteoporosis |
| Pigment gallstones |
| Reticulocytosis |
| Thrombocytosis |
| 5 – 29% of people have these symptoms: |
| Abnormality of the nervous system |
| Cholestasis |
| Elevated serum creatinine |
| Hypoxemia |
| Increased LDH activity |
| Persistence of hemoglobin F |
| Unconjugated hyperbilirubinemia |
| 1 – 4% of people have these symptoms: |
| Increased mean corpuscular volume |
| Microcytic anemia |
| Additional symptoms that recipients may experience: |
| Abdominal pain |
| Cardiomegaly |
| Hematuria |
| Hepatomegaly |
| Hypertension |
| Jaundice |
| Priapism |
| Renal insufficiency |
| Retinopathy |
| Splenomegaly |
| Stroke |
The table above contains information collected from the Human Phenotype Ontology (HPO) database.
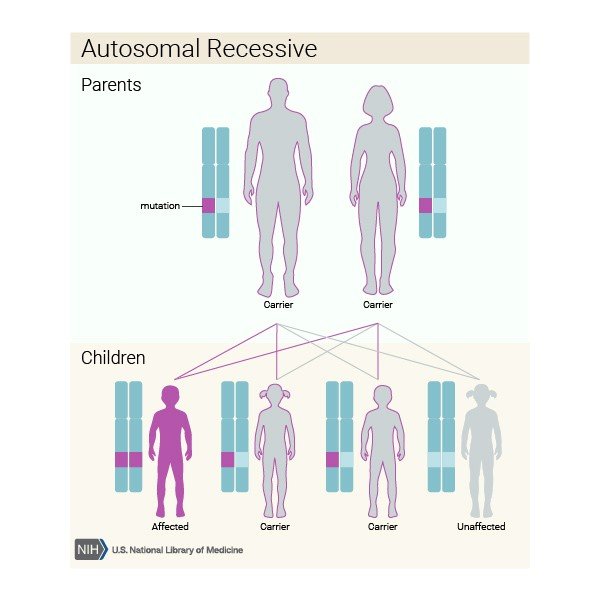
Sickle cell disease is inherited in an autosomal, recessive pattern. A person who carries one copy of the mutated gene is said to be a carrier of the sickle cell trait but will not exhibit symptoms of sickle cell disease. If two people are both carriers for the sickle cell trait and have a child, there is a 25% chance that this child will have SCD, a 50% chance of being a carrier, and a 25% chance that the child will not be a carrier nor have SCD. Reference Graphic 2. Inheritance of Sickle Cell Disease below for a depiction of this description:
Graphic 2. Inheritance of Sickle Cell Disease
1 Genetics Home Reference: Your Guide to Understanding Genetic Conditions. 2020. Sickle Cell Disease. [online] Available at: < https://ghr.nlm.nih.gov/condition/sickle-cell-disease > .
2 Genetics Home Reference: Your Guide to Understanding Genetic Conditions. 2020. Sickle Cell Disease. [online] Available at: < https://ghr.nlm.nih.gov/condition/sickle-cell-disease >.
Need more help with this?
Don’t hesitate to contact us here.