
Frequently asked questions
Error message: Sorted sam/bam file is required
The system expects the BAM file to be sorted by the coordinates so that it can be processed efficiently. If the BAM file is not sorted, the system will display the error message “Sorted sam/bam file is required”. A BAM file can be sorted using the samtools program.
The source code for samtools is available here. For Microsoft Windows users, there is an executable version available for download here. Please note that also the Windows version needs to be run from the command prompt.
The command below will take the input file file original.bam, sort it, and output the results to sorted.bam. The sorting process uses temporary files on disk and it requires as much free disk space as is the input file size.
Error message: “No track name provided” for a BAM file
An error message “No track name provided” for a BAM file actually indicates that there is an issue with the BED file containing multiple tracks, and the system does not know which one of them to use for performing the read depth analysis.
The fix is to remove extra tracks from the BED file so that it contains only a single track and then to upload the fixed BED file, and then to re-upload the same BAM file again to rerun the BAM analysis.
Uploading multiple fastq.gz files
To upload multiple fastq.qz files, you can select multiple files from the file selection window that opens. All the files have to be selected together, they can not be selected separately, as subsequent uploads will overwrite previously uploaded files.
1. In the Result Submission (stage 2) phase, click the Choose Files button next to the text “Gzipped fastq files”.

2. In the file selector dialog window that opens, you can select multiple files by clicking on them while keeping the Control button pressed (Command button ⌘ on macOS).
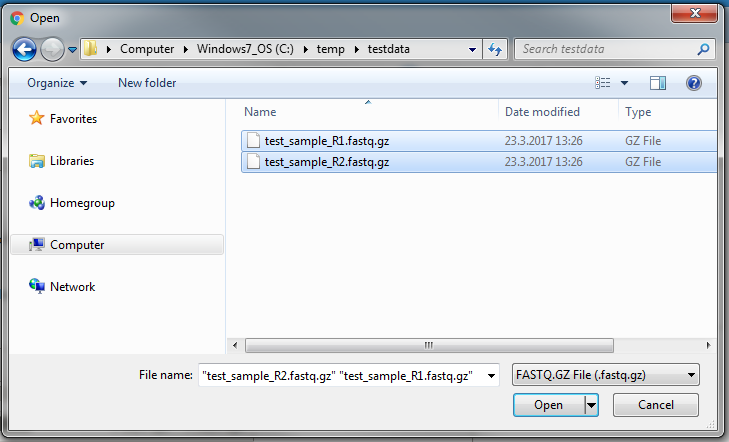
Note that the “File name” text box indicates multiple files having been selected.
Click Open in the file select dialog after you have selected all files that you want to upload.
3. Observe that next to the “Choose Files” button the number of selected files is shown, and also the progress bar below it indicates that multiple files are being uploaded, displaying the upload progress for each.

4. Wait until it the “Done” is displayed in the progress bar.

5. Wait until the “Processing status” display for “FASTQ” indicated “Succeeded”. Please note that there is only one processing row in the processing status table for FASTQ, even if multiple files have been uploaded (they are all processed in one go).

Are the only variants in my VCF to be considered those which are unfiltered?
Answer: Yes, only variants that have the filter set either to ‘PASS’ (Not Filtered), or to ‘.’ (no data, filtering not performed) will be considered in the assessment. Any other value, such as ‘LowQual’, ‘LQ’, etc. means that a quality filter has been applied to exclude the variant according to VCF format convention.
In my laboratory we also analyse filtered variants, but the EQA does not consider them for assessment, what shall I do?
Solution. You can set the FILTER column to PASS for all the variant calls that you wish to be included in the variant concordance assessment.
The VCF I am trying to submit is rejected by the submission platform, what is going on?
Answer: The VCF that was submitted could not be understood by the EQA process.
- First, be sure you have not tried to modify the VCF output using Excel. Although the VCF files can be opened in Microsoft Excel as tab-separated files, saving them from Excel introduces extra quote characters in the file, making it invalid.
- Chromosome naming should be either “1”, “2”, … “X”, “Y”, “MT”, or “chr1”, “chr2”, … “chrX”, “chrY”, “chrM”.
Solution: Provide unedited, native VCF files as they are output from your pipeline. Please contact our support if you think that the VCF is correct, but nevertheless rejected. VCF files can be correct in many different ways and the EQA process might not recognise them all, and a fix can be introduced quickly.
Where can I find more information about the VCF file format?
Answer: The Broad Institute has a nice page about this here.
Why do I need to provide a BED file?
Answer:
- The BED file provides the exact information about the area of the genome that is covered by the kit/test used for the test. If the BED file is not provided, the system will assume that the region of interest is the whole exome. As a consequence, certain quality metrics will show abnormally low values (e.g. average read depth).
How can I specify that my region of interest (the genes I am testing) is only a subset of the region captured by the kit/method used for the test?
Answer: The upload system requires you to provide the target of your test using a BED file. If you need to specify that only a subset of the captured target region is analysed, you can provide a list of genes to include. It is also possible to perform the complementary operation, that is to exclude genes using a list of HGNC gene names.
What software can I use to compress or decompress files using the gzip format (file extension .gz) on Windows?
Answer: Note that the gzip (.gz) format is different from the zip (.zip) file format. To work with gzip files on Windows, you need to use a file compression/uncompression application which supports that format. Some freely available alternatives include:
- 7-zip (http://www.7-zip.org/), a file archiver program which supports many different formats, including gzip. Free of charge and open source. There is also an alternative installer available which does not require Administrator privileges at PortableApps.com.
- gzip.exe, a command line executable, available for Windows here. This is a program that can be used from within the Windows Command Prompt.
How do I compress a .fastq file into a .fastq.gz file on Windows?
Answer: See above for a list of programs that can be used to work with gzip files. The following instructions assume you are using 7-zip. If you are using some other program, please see its’ user guide.
- Make sure you have 7-zip installed.
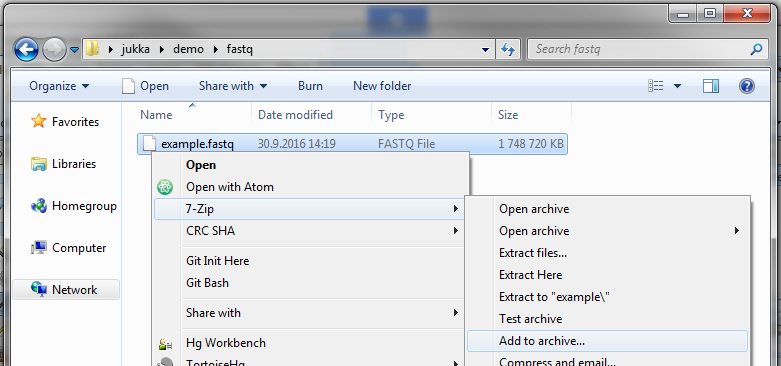
- Open the folder containing your .fastq file in Windows Explorer, right-click the .fastq file and select 7-Zip > Add to archive…
![]()
- From the Add to Archive Window, select Archive format: gzip.
![]()
- Click OK.

Done. You should now have a .fastq.gz file in the same folder as the orginal .fastq file.
Can I submit a gVCF (genomic VCF) file?
Yes, gVCF (“genomic VCF”) files can be submitted. They are regular VCF files, but contain some extra information in the form of non-variant blocks (see for example this GATK article for a more detailed explanation). These files are fine, as they are handled as regular VCF files.
Post your comment on this topic.